Što je Marfanov sindrom?
Marfanov sindrom opisuje složeni nasljedni poremećaj vezivnog tkiva koji uglavnom pogađa oči, kardiovaskularni sustav i sustav skeletnih mišića. Međutim, s obzirom da je svaki organ sastavljen od vezivnog tkiva, Marfanov sindrom idealno može uništiti i jako ometati rast i funkciju svakog anatomskog mjesta.
Sindrom se prenosi kao autosomno dominantna osobina: stoga smo suočeni s ozbiljnom genetskom bolešću, koja ima izrazito varijabilnu fenotipsku ekspresiju (defekti se mogu jako razlikovati od obitelji do obitelji ili od pacijenta do pacijenta).
Ono što potiče Marfanov sindrom je promjena gena FBN1 (na kromosomu 15), koji kodira fibrilin-1, vrlo važan vezni glikoprotein koji čini strukturnu potporu mikrofibrilima.
Mikrofibrile: koje se sastoje od fibrilina, mikrofibrile su prisutne u izvanstaničnom matriksu, u kojem tvore mrežu za taloženje elastina u elastičnim vlaknima. Iako je sveprisutna u tijelu, mikrofibrile obiluju uglavnom u aorti, ligamentima i zonulama cilijarnih tijela (na razini oka).
Kao autosomno dominantna bolest, samo djeca koja su naslijedila gen za FBN-1 promijenjen od strane oba roditelja, pogođena je Marfanovim sindromom. Ipak, u jednom od 4 slučaja bolest je posljedica spontanih mutacija u bolesnika koji nemaju obiteljsku anamnezu.
Ime bolesti dolazi od francuskog pedijatra koji ju je prvi put opisao 1896. godine (A. Marfan), nakon čega je morao čekati do 1991. da identificira izmijenjeni gen uključen u simptomatsku manifestaciju: otkrivač je bio F. Ramirez.
Pogledajte Video
X Pogledajte videozapis na youtubeuuzroci
Spomenuli smo da je Marfanov sindrom neposredan izraz mutacije gena koji kodira fibrilin-1. Pokušajmo produbiti temu kako bismo razumjeli koji je mehanizam uspostavljen za pokretanje sindroma.
FIBRILLINA 1 je glikoproteinska komponenta elastina, neophodna za osiguranje i održavanje elastičnosti i čvrstoće tkiva. U fiziološkim uvjetima, fibrilin 1 se veže na drugi protein, poznat kao TGF-beta (ili transformirajući beta-faktor rasta). Čini se da je TGF-beta uključen u štetne procese koji pogađaju glatke mišiće krvnih žila i izvanstanični matriks. Polazeći od tih pretpostavki, neki autori su uvjereni da je Marfanov sindrom, osim mutacije gena FBN-1, posljedica i viška TGF-beta, posebno u aorti, u srčanim zaliscima i plućima.
učestalost
Procjenjuje se da Marfanov sindrom utječe na 1 subjekt na svakih 3.000-5.000 rođenih i manifestira se nejasno između muškaraca i žena, bez sklonosti za preciznu pasminu. Statistike pokazuju da 75% pacijenata ima pozitivnu obiteljsku povijest; u preostalih 25% uzrok leži u sporadičnim mutacijama koje se na neki način povezuju s uznapredovalom dobi oca u vrijeme začeća.
Djeca s ekstremno teškim oblicima Marfanovog sindroma imaju očekivano trajanje života kraće od jedne godine.
Prije evolucije kirurških strategija na otvorenom srcu, većina pacijenata s Marfanovim sindromom imala je prosječan životni vijek od 32 godine; zahvaljujući stalnom poboljšanju medicinskih i farmakoloških terapija, danas pacijenti s Marfanovim sindromom žive u prosjeku do 60 godina.
Znakovi i simptomi
Za više informacija: Simptomi Marfanov sindrom
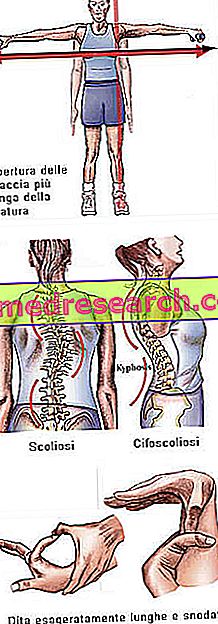
Marfanov sindrom može raditi potpuno asimptomatski. Oboljeli pacijenti imaju izrazito vitku strukturu, koja je nerazmjerno visoka i mršava. Donji i gornji udovi imaju mnogo veću dužinu od debla (dolikostenomegalija). Također se govori o arachnodactyly kako bi se najbolje izrazio koncept pretjerane duljine prstiju, tipičan za subjekte koji su pogođeni Marfanovim sindromom: ruke su stoga uspoređene s nogama pauka.
Što se tiče visine, ovi pacijenti imaju prosječnu visinu iznad 97. percentila.
Ostale karakteristike koje se često nalaze u bolesnika s Marfanovim sindromom uključuju:
- Ruke otvorene veće od visine
- Loose zglobovi → pretjerana pokretljivost zglobova
- Deformacija prsnog koša
- Kristalno premještanje
- Gornji dio tijela manje razvijen od donjeg dijela
- Spontani pneumotoraks (11%)
- skolioza
- Strije u koži na bedru, leđima, deltoidnoj, prsnoj razini
Među najproblematičnijim znakovima povezanim s Marfanovim sindromom, spominjemo prolaps srčanog zaliska i insuficijenciju mitralne valvule: slično stanje može lako potaknuti dilataciju aortnog prstena i disekcije aorte.
Tablica pokazuje znakove koji se mogu naći u bolesnika s Marfanovim sindromom. Opisani likovi nisu uvijek prisutni, ali se dobar dio njih može pronaći.
| Pogođena anatomska lokacija | Mogući simptomi |
sladak | Strije u torakalnom, lumbalnom i sakralnom području |
oči | Oštećenje vida, astigmatizam, odvajanje retine, uski kut glaukoma, dislokacija leće, kratkovidost |
Struktura kostiju | Artralgija, kifoskolioza, dolicostenomelija (prekomjerni razvoj dužine ekstremiteta u odnosu na trup), hipermobilnost, visoko nepce, deformirana prsa, ravne noge, uski i tanki zapešća, abnormalni povratak / izbočenje prsne kosti, skolioza, zakrivljena ramena, spondilolisteza |
Dita | arachnodactyly |
pluća | Spontani pneumotoraks, dispneja, idiopatska plućna opstruktivna bolest |
Promjene lica | Ogivalno nepce (malformacija nepca), mandibularna retrognatija (defekt u razvoju čeljusti), izduženo lice |
srce | Angina pektoris, aneurizma abdominalne aorte, srčana aritmija, dilatacija / disekcija torakalne aorte, aortna insuficijencija, prolaps mitralne |
jezik | Težina jezika |
dijagnoza
S obzirom na više od 200 mogućih mutacija, uporaba genetskih markera gotovo je nemoguća za dijagnostičke svrhe.
Utvrđivanje Marfanovog sindroma nije uvijek tako neposredno, s obzirom na to da fenotipska ekspresija mutacije nije uvijek očita i jednostavna za identifikaciju. Dijagnostičko kašnjenje može ozbiljno ugroziti opstanak pacijenta: samo zamislite, na primjer, neuspjeh u prepoznavanju kardiovaskularnog problema.
Dijagnostički kriteriji za Marfanov sindrom izrađeni su na međunarodnoj razini 1996. godine: dijagnoza se sastoji u istraživanju obiteljske anamneze povezane s kombinacijom glavnih i manjih pokazatelja sindroma.
Neki od mnogih korištenih dijagnostičkih testova su:
- echocardiogram
- Magnetska angiorezonancija i CT (za ispitivanje aorte)
- magnetska rezonancijska angiografija (MRA) s kontrastnom tekućinom (kako bi se utvrdile unutarnje strukture aorte)
- ispitivanje s prorezanim svjetiljkama (za analizu mogućeg pomjeranja leće)
- mjerenje očnog tlaka (kako bi se naglasila moguća prisutnost glaukoma)
- genetski testovi (preporuča se prije zasnivanja djeteta da se utvrdi sindrom ili ne)
terapije
Budući da je genetska bolest, ne postoji lijek ili liječenje koje može preokrenuti bolest.
Upotreba lijekova je ipak bitna za ublažavanje simptoma i izbjegavanje bilo kakvih komplikacija, posebice srčanih. U tu svrhu posebno su prikladni lijekovi za smanjenje krvnog tlaka, kao što su sartani (posebno), ACE inhibitori i beta-blokatori.
U kontekstu Marfanovog sindroma, pacijenti koji također pate od skolioze mogu slijediti specifičnu skrb, kao i za one koji su pogođeni glaukomom.
Kirurgija je zamisliva kako bi se ispravila anomalna dilatacija aorte, element koji često ujedinjuje većinu bolesnika s Marfanovim sindromom.
Nastavak: Marfanov sindrom - droga i njega »


